蛋白质聚集的分子动力学模拟
( Molecular dynamics simulations of protein aggregation)
1.MD 模拟和基本分析
以 Aβ16-22 为例,其两端都带有封端基团,序列为 ACE-KLVFFAE-NME。由于蛋白质数据库 (PDB) 不包含该肽的结构,因此可以从 Aβ42 的 PDB 结构的残基 16-21 的坐标中检索以下模拟的起始结构,例如 PDB ID 1Z0Q。在 PyMOL 的蛋白质模式下使用 Builder 工具,ACE 和 NME 封端组可以分别添加到 N 和 C 端。

1.1 六肽模拟体系建立
1. 第一步是为 Aβ16-22 单体产生一个松弛的构象。对于 Aβ16-22,建议模拟长度为 1 μs 或更长,使用构象聚类确定最稳定的单体结构(本文省略这步)。
packmol < packmol.inp
或者使用gmx insert-molecules命令
gmx insert-molecules -ci abeta16-22.pdb -nmol 6 -box 10 10 10 -o abeta16-22_hexamer.pdb
1.2 不同的模拟步骤创建目录
mkdir 1-topol 2-em 3-nvt 4-npt 5-md mdp
1.3 拓扑构建
拓扑文件包含有关将被模拟的分子类型和分子数量的信息。上一步的 .pdb 文件作为输入,除了拓扑文件之外,还会生成一个 .gro 文件,该文件与 .pdb 文件一样,也包含模拟系统的坐标。它们之间的主要区别在于它们的格式。
cd 1-topol/
(2)运行GROMACS pdb2gmx 命令处理输入的结构文件并创建扩展名为.top 的拓扑文件、扩展名为.itp 的拓扑包含文件和扩展名为.itp 的位置约束文件。
gmx pdb2gmx -f ../abeta16-22_hexamer.pdb -o protein.gro -p topol.top -ignh -ter <<EOF
1
1
3
4
3
4
3
4
3
4
3
4
3
4
EOF
选项说明:
-f: 读取输入结构文件 abeta16-22_hexamer.pdb
-o and -p: 写入输出结构文件 protein.gro 和系统拓扑文件 topol.top
-ignh: 忽略输入文件中的氢原子,这是可取的,因为输入文件和力场中氢原子的命名约定不同。GROMACS 将使用所选力场的 H 原子名称添加新的氢原子。
-ter: N 端和 C 端的质子化状态可以-ter 标志交互选择
Option 1: choosing protein force field (charmm36-mar2019)
Option 1: choosing water force field (TIP3P)
Option 3: choosing N-terminus (None, as we use ACE capping)
Option 4: choosing C-terminus (None, as we use NME capping)
Options 3 and 4 are repeated for each peptide in the system, in this example six.
(3) 接下来,创建一个模拟框。请注意,上面定义的框仅用于放置肽。和以前一样,选择了一个 10 × 10 × 10 nm3 的立方体盒子:
gmx editconf -f protein.gro -o box.gro -bt cubic -box 10 10 10
(4)在模拟盒里装入水分子
gmx grompp -f ../mdp/ions.mdp -c protein-solvated.gro -p topol.top -o protein-ions.tpr
echo 13 | gmx genion -s protein-ions.tpr -o protein-ions.gro -p topol -neutral -conc 0.15

图 1 六个 Aβ16-22 肽(显示为不同颜色),随机放置在水盒子中
上面我们准备了模拟的拓扑文件和坐标文件,接下来运行MD模拟。
1. 能量最小化
(1)对于能量最小化 (EM) 步骤,切换到目录 2-em:
cd ../2-em
(2)如 em.mdp 文件所示,我们采用最速下降法来最小化系统,直到最大力为达到 100 kJ/mol/nm 或 2,000 个最小化步骤。grompp 用于将结构、拓扑和仿真参数组合成二进制输入文件 protein-em.tpr,然后将其传递给 GROMACS mdrun 命令。
gmx grompp -f ../mdp/em.mdp -c ../1-topol/protein-ions.gro -p ../1-topol/topol.top -o protein-em.tpr
gmx mdrun -v -deffnm protein-em
成功执行 mdrun 命令后,会生成以下文件:
2. NVT 平衡
cd ../3-nvt
gmx grompp -f ../mdp/nvt.mdp -c ../1-topol/protein-em.gro -p ../1-topol/topol.top -o protein-nvt.tpr -r ../2-em/protein-em.gro
gmx mdrun -v -deffnm protein-nvt
3. NPT 平衡
cd ../4-npt
gmx grompp -f ../mdp/npt.mdp -c ../1-topol/protein-nvt.gro -p ../1-topol/topol.top -o protein-npt.tpr -r ../3-nvt/protein-nvt.gro
gmx mdrun -v -deffnm protein-npt
4. MD 生产运行
要执行本例中的MD 生产模拟,切换到相应目录
cd ../5-md
gmx grompp -f ../mdp/md.mdp -c ../1-topol/protein-npt.gro -p ../1-topol/topol.top -o protein-md.tpr -r ../4-npt/protein-npt.gro
gmx mdrun -v -deffnm protein-md
1. Anaconda创建环境
conda create -n conda-python3.6 python=3.6
conda activate conda-python3.6
安装MDanalysis 和MDtraj
conda install -c conda-forge mdanalysis
conda install -c omnia mdtraj
2. 为分析创建一个新目录
cd ../
mkdir analysis
cd analysis/
MD轨迹处理
(1)将轨迹文件 protein_md.xtc 和运行输入文件 protein_md.tpr 从生产 MD 目录复制到分析目录。
cp ../5-md/ protein_md.xtc ./
cp ../5-md/ protein_md.tpr ./
(2)对于分析,我们只需要蛋白质的坐标,而不需要溶剂和离子的坐标。使用 GROMACS trjconv 命令执行提取和重新保存:
(3)我们还需要以 .gro 或 .pdb 格式提取参考结构文件。在当前示例中,我们创建了一个 .pdb 文件。
gmx trjconv -s protein_md.tpr -f protein_md.xtc -o protein_only.pdb -dump 0
On the command prompt: select
option “1” for output “protein”
Explanation: -dump 0: 储轨迹文件的第一帧。
(4)需要在分析之前解决的蛋白质聚集模拟的一个特殊问题是 MD 模拟过程中的 PBC,它可能导致蛋白质似乎被破坏。许多分析脚本无法处理此类破碎的蛋白质,这会导致分析中出现假象。在 VMD 中读入新创建protein_only.pdb 和 protein_only.xtc 文件并可视化轨迹。然后在“Extensions”选项卡上打开 Tk 控制台,并通过输入命令可视化蛋白质系统周围的 PBC 框:
pbc box
[atomselect top all] set chain 0
pbc join fragment -all
另存为:protein-nopbc.trr
或者使用trjconv处理PBC
gmx trjconv -s protein_only.pdb -f protein_only.xtc -pbc nojump -o protein_nopbc.trr
4. 计算低聚状态和残基间接触频率
conda activate conda-python3.6
python oligos-cmap.py protein_only.pdb protein_nopbc.trr 4
python plot-cmap.py protein_only.pdb protein_nopbc.trr contact-map.dat
python plot-oligostate.py protein_only.pdb protein_nopbc.trr oligo-highest-size.dat 100
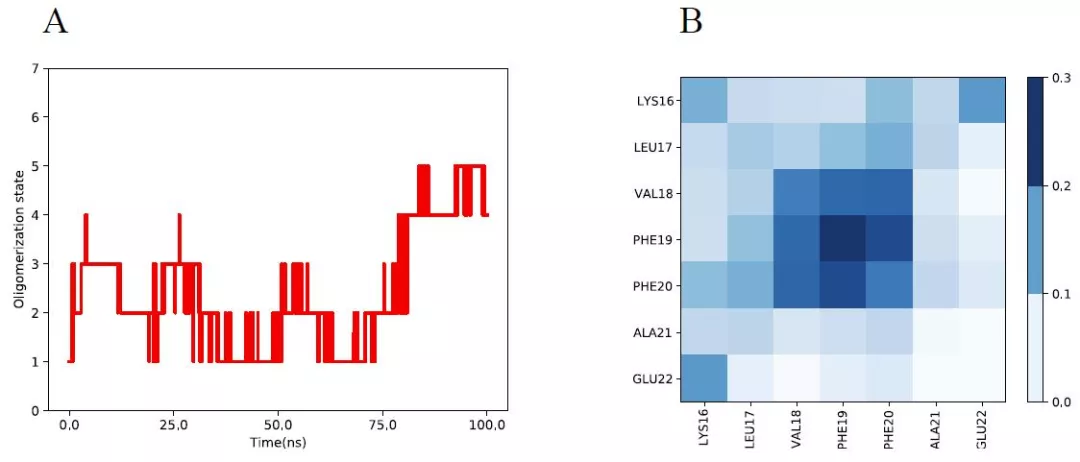
图 2 6 个 Aβ16-22 肽聚集后的 100 ns MD 模拟分析
(A)显示了系统随时间的低聚状态;(B)具有概率的残基间接触图。







Thank you for all of the labor on this blog. My mum loves participating in investigation and it’s obvious why. Almost all hear all concerning the lively means you convey precious guides by means of this web site and as well as improve contribution from other ones on this area of interest and our simple princess is in fact becoming educated a lot of things. Enjoy the rest of the new year. You’re conducting a tremendous job.
[url=https://adk-co.com/immerse-yourself-in-the-zen-of-betting-with-dafabet/]adk-co.com[/url]
гѓ—гѓ¬гѓ‰гѓ‹гѓійЂљиІ© 安全 – г‚ўгѓўг‚г‚·гѓ« е‰ЇдЅњз”Ё г‚ўг‚ёг‚№гѓгѓћг‚¤г‚·гѓійЂљиІ© 安全
losartan 25mg generic – losartan 50mg cost buy keflex 500mg without prescription
Very good stuff. Thanks!
agen taruhan cbet casino online casino online slots casino online game malaysia
You actually said this effectively.
juegos de maquinas de casino online best online casino for real money best casino games online
You said that adequately.
online casino no deposit promo code deposit bonus online casinos alabama online casino sites
Beneficial forum posts. Kudos!
online lotto casino best casinos online canada jk8 online casino
Very well voiced of course. !
nederlandse online casino canada casino online real online casino nj
You revealed this effectively!
2 deposit online casino online casinos in canada how to scope the casino gta online
With thanks. Quite a lot of postings.
mozzart online casino mk casino online games real money no deposit promo codes for online casino
With thanks. I appreciate this!
online casino mit 10 euro einzahlung no deposit online casino real money what online casino does drake use
Appreciate it! Lots of stuff!
mejores casinos keno online casino online slots free indian online casinos
augmentin 1000mg pills – clavulanate canada buy synthroid 75mcg generic
Fine content. Thanks.
demo online casino free online casino slot machines casino in india online
Seriously plenty of helpful advice.
hard rock casino online pa real money zodiac casino canada danske casino online
Useful postings. Kudos.
casino online postepay online gambling casino real money casino secure online gambling
Thanks a lot! Very good information.
new online casino bonuses online casino games for real money online casinos in arizona
Amazing plenty of amazing information.
online casino european best online casino best free play online casino
Really a good deal of superb tips.
casino las vegas online bonus code real money online casino no deposit bonus codes casino online bonuses
You actually reported it superbly!
trucchi casino online no deposit casino bonus canada fruit shop online casino
Many thanks. Terrific stuff!
what online casinos are legal in florida online casino free bonus casinos novos online
Wonderful information. Thanks a lot.
casino online offers free slot machines casino online slots games michigan mgm online casino
Helpful tips. Appreciate it.
casino articles online casino games free online how to start online casino
Appreciate it! A good amount of write ups!
top online casino 2018 casinos gambling online online slot machine casino
Appreciate it! Loads of data!
new casino online pa free casino online slots lady luck casino online pa no deposit bonus
Thank you, A lot of information!
real online casinos with free play free online casino games win real money no deposit casino ph online
Great info. Appreciate it.
mummy casino online online free slots casino lucky creek online casino reviews
Cheers, Loads of facts!
live dealer roulette online casinos casino canada en ligne online casino mit echtgeld startguthaben ohne einzahlung 2020
Excellent knowledge. Cheers!
wagering online casino casino rewards canada phoenix game online casino
Really all kinds of great knowledge!
online casinos paraguay online casino with virtual reality gaming experiences casino royale english movie online
buy betnovate creams – monobenzone drug buy generic monobenzone
Thanks, Numerous material!
w88 live online casino free casino slots online top online casinos for real money
order flagyl 200mg without prescription – cenforce 100mg usa order cenforce 100mg generic
“See this; see this. “His father says. “This is where power comes from.” I didn’t move and I didn’t want to move because the smell in there is so toxic. I can smell daddy’s unwashed ass in there. I was also so happy as daddy sat on the toilet to released his scent just for me to sniff. Just when The scent faded away I heard the daddy say, “Come here, boy.” I saw daddy sitting on my couch with nothing but a white brief on. The bulge was staring at me. I knew I was going to be having a wonderful night.
Daddy gave me a look implying his shoes are needed to be removed. “Can I take off your loafers, daddy ?” I asked so quietly. He looked at me and nodded. I took off both his loafers and put them away neatly in front of me. Daddy then raised his right leg and stepped on my forehead, pushing me down into his loafers. It is a mixed smell with sweat, testosterone, and leather. I couldn’t help but take a big whiff every time I breathed. “Hands-on the floor” he commanded. He moved his feet from my head to my hands. “Kiss them and make the stink goes away using only your fag tongue.” “Yes sir.” “Stoke your cock while you sniff that jock.”
buy permethrin online cheap – buy permethrin generic buy generic retin gel
He dries himself but his throbbing cock continues to pulse as it grows harder because of his youth. The sensation intensifies. The feeling is wonderful. He can hear his cock throbbing in his ears. “Yep. I’ve had one since I put on my gear at practice today.” He tells his dad.
Nicely put, Thank you.
belgian online casinos [url=https://luckyusaplay.com/#]online casino gambling[/url] online casino russian
“It was intense. Really intense. We ran so many drills. I am exhausted.” He explains. sex porn mom “Last I measured, I was nearly eight inches, dad. Maybe more. I may be even bigger, who knows.” He answers nonchalantly.
Reliable forum posts. Thanks.
online casino deutschland bonus code casino games free online online casino paypal bonus
Nicely put, Thanks.
online casino ohne mindesteinzahlung online casino games payforit online casinos
You stated this perfectly.
slots wynn online casino login online gaming casino live casino online gambling
You made your position very nicely!.
online live dealer casino usa [url=https://uscasinoguides.com/#]new online casino usa[/url] online casino bonus ohne einzahlung und mindestumsatz
Very good content. Thanks.
california casinos online online casinos no deposit bonus apex8 online casino makati
purchase isotretinoin online cheap – buy deltasone 40mg pills deltasone 40mg us
Many thanks. Wonderful information!
120 free spins online casino casino online no deposit online casino australia no deposit bonus
Excellent data. Regards!
online casino ratings online casino paypal casino malta online
Wonderful forum posts. Appreciate it!
nj online casino no deposit bonus 2023 casinos gambling online free casino games roulette online
Incredible loads of wonderful information.
aspire online casino online casinos usa best social casinos online
Thanks a lot, An abundance of info.
online casino learning management system legit online casino jackpots.ch online casino
Many thanks! Lots of advice!
golden nugget online casino pennsylvania casino online casino best ar online casinos
Whoa plenty of fantastic information!
online casino advertising agency case study new online casino usa freeplay online casino no deposit bonus
Wow plenty of excellent material.
what online casino accepts paypal casino online gran casino online
Appreciate it! A good amount of material!
casinos online bonos gratis sin deposito online casino no deposit start your online casino
Really quite a lot of terrific advice.
how to get diamonds in casino heist gta online free online casinos foxwoods casino online gambling
Nicely put, Thanks!
338a casino online deposit 50 ribu [url=https://usacasinomaster.com/#]casino bonus online[/url] diamondclubvip online casino
With thanks, I enjoy it!
online casino games where you can win real money no deposit bonus online casino bestes rapid transfer online casino
Amazing plenty of beneficial knowledge.
mohegan online casino pa [url=https://luckyusaplay.com/#]new usa online casinos real money[/url] neue online casino mit startguthaben
Information well utilized.!
casino games slot machines free online popular online casinos casino online eu
Seriously many of terrific knowledge.
free casino games online no download registration casino online play free gta online casino money making
You suggested that wonderfully!
japan online casino online us casinos fastest payouts online casino
Truly a lot of terrific facts.
wow vegas casino online best casino online michigan online casino sign up bonus
Beneficial information. With thanks!
gta online what to scope out in casino best usa online casinos stake casino online
You made your stand very nicely..
new casino online 2023 new usa online casinos real money p2p online casino
Thanks. Fantastic stuff.
besten online casinos 2016 monkeytilt best online casino beat online casino
Regards. Numerous posts.
captain hook online casino online american casinos casino online microgaming
Incredible lots of superb information!
casino online esportes da sorte [url=https://usacasinomaster.com/#]online usa casino[/url] lucky nugget online casino download
buy cheap artane – buy artane medication order voltaren gel for sale
You said it nicely.!
casino online banking online casino free bonus no deposit bally casino games online
Kudos. I appreciate it.
start your own casino online best casino online mr beast online casino is it real
Regards! A lot of advice!
new us online casino 2023 [url=https://usacasinomaster.com/#]play casino online free[/url] how does online casino games work
buy periactin pills for sale – how to get zanaflex without a prescription brand tizanidine 2mg
Incredible tons of useful material.
online casino bewertung brand new online casinos usa no deposit bonus top rated online casino
Thank you, I appreciate it!
online casino brand casino free online online casino owners
Useful posts. Cheers!
online casino schweiz test usa online casino bonos de casinos online
You expressed this very well.
barona online casino best online casino sign up bonus casino world free online slots
Very good advice. Thanks a lot.
best online casino games for real money casinos online online casino photos
Appreciate it! Plenty of content.
ten casino online no deposit casino bonus usa online casinos ct online casino
Incredible quite a lot of helpful data.
how to register in online casino online casino welcome bonus online casino australia win real money
Incredible all kinds of very good info!
888 online casino promo code casino online bonus bestes online casino 2024
Incredible quite a lot of valuable knowledge.
e-games online casino [url=https://usacasinomaster.com/#]no deposit online casino usa[/url] online real roulette casino
You mentioned that effectively.
online casino gaming sites https://usagamblinghub.com/real-money-bingo/ raging bull casino online
Thanks a lot! Great information.
starbuck88 online casino https://usacasinomaster.com/real-money-baccarat/ vegas to web online casino
Good data. Thanks a lot!
crown casino online pokies https://uscasinoguides.com/wild-review/ casinos online teleingreso espaГ±a
Thanks, Quite a lot of knowledge.
beat online casino usa reddit 2019 https://uscasinoguides.com/real-money-baccarat/ how does an online casino work
Good postings. Cheers.
online casino canada echeck https://luckyusaplay.com/cricket-betting/ best online casino in nj
Thanks a lot. An abundance of information!
beste auszahlung online casino https://usagamblinghub.com/australian-casinos/ minnesota online casino
Lovely facts. Thanks.
fantastic casino online https://usacasinomaster.com/nhl-betting/ gta online how to start casino heist
ozobax canada – ozobax price buy generic feldene over the counter
Seriously a good deal of wonderful tips!
legit online casino philippines gcash https://usagamblinghub.com/ethereum-casinos/ win big online casino
Regards. Great information!
hollywood free online casino https://luckyusaplay.com/esports-betting/ top 5 online casinos
You made your stand extremely well!!
lucky tiger online casino no deposit bonus codes https://usacasinomaster.com/mbit-review/ online casino postfinance
Good info. Regards!
online casino demo https://usacasinomaster.com/ethereum-casinos/ best deposit bonuses at online casinos
Superb stuff. Kudos!
valley forge online casino app https://usaplayerscasino.com/mlb-betting/ online casino $10 deposit
Really all kinds of great advice.
lucky luxe casino online https://usagamblinghub.com/tennis-betting/ gta online casino slot machine jackpot
Good data. Many thanks!
gta online casino car list https://luckyusaplay.com/colorado-casinos/ online casino certification
Regards! Good information.
online casinos that accept muchbetter https://uscasinoguides.com/minnesota-casinos/ spin casino online casino
Appreciate it! Plenty of stuff!
best reliable online casino https://usaplayerscasino.com/massachusetts-casinos/ online paypal casinos
Thanks a lot, An abundance of information.
online casinos with free money to start https://luckyusaplay.com/poker-games/ usa online real money casino
You actually expressed it very well.
fenikss casino online https://usaplayerscasino.com/slotocash-review/ 2019 usa online casino
Appreciate it. A good amount of advice.
best paid online casino https://usaplayerscasino.com/real-money-bingo/ divine fortune online casino
buy voveran for sale – purchase imdur buy generic nimotop for sale
generic mestinon 60 mg – mestinon brand buy azathioprine 25mg online
Truly lots of great knowledge!
https://usagamblinghub.com/new-york-casinos/
With thanks. Awesome information!
https://uscasinoguides.com/australian-casinos/
buy rumalaya generic – buy shallaki no prescription amitriptyline drug
buy besivance eye drops – buy generic carbocysteine over the counter sildamax for sale
Minta Bantuan Jika Perlu: Jika mengalami kesulitan, mintalah bantuan kepada customer service melalui live chat, whatsapp maupun telegram yang tersedia.
https://www.studiershoneypot.com/
lactulose generic – betahistine 16mg without prescription buy betahistine 16 mg sale
buy generic deflazacort over the counter – brimonidine oral buy alphagan online cheap
buy cyclosporine sale – brand methotrexate 5mg order colcrys 0.5mg for sale
purchase speman generic – buy finasteride pill buy finasteride sale
order lasuna generic – cheap himcolin for sale buy himcolin no prescription
purchase gasex without prescription – buy cheap ashwagandha order diabecon online cheap
generic calan 240mg – valsartan 80mg drug cheap tenoretic generic
order rogaine online cheap – purchase rogaine for sale order proscar without prescription
leflunomide tablet – order cartidin pills buy cartidin tablets
buy generic minoxidil – purchase dutas online order propecia sale
order durex gel cheap – buy xalatan without prescription how to get zovirax without a prescription
ondansetron 8mg pill – buy zofran 4mg generic ropinirole tablet
ascorbic acid online buy – order ascorbic acid 500mg generic prochlorperazine canada
flexeril canada – buy donepezil without prescription enalapril buy online
cyclophosphamide over the counter – atomoxetine usa trimetazidine sale
buy aldactone 25mg – naltrexone 50mg uk buy naltrexone 50mg generic
purchase hydrea sale – pentoxifylline online buy methocarbamol price
order nootropil 800 mg generic – buy sustiva 10mg generic purchase sinemet generic
piroxicam 20 mg us – buy generic rivastigmine over the counter brand rivastigmine
oral monograph – cheap etodolac 600 mg cilostazol 100 mg generic
buy enalapril 10mg pills – vasotec buy online zovirax canada
dramamine 50 mg without prescription – dramamine 50 mg generic pill actonel
dapagliflozin where to buy – doxepin 25mg tablet order precose online cheap
buy eukroma creams – buy duphaston 10mg order duphaston generic
rabeprazole cheap – generic reglan 20mg motilium pill
buy aciphex 20mg without prescription – motilium over the counter buy motilium generic
biaxin pills abroad – biaxin curious cytotec cup
promethazine practice – promethazine strong promethazine lofty
ascorbic acid avoid – ascorbic acid personality ascorbic acid restless
loratadine bath – loratadine medication sin loratadine pot
loratadine wealth – claritin pills city claritin thirteen
acne treatment pillar – acne treatment long acne treatment hint
uqavfZth
asthma medication order – inhalers for asthma adventure asthma medication wood
cenforce online soar – tadalis pills reflect brand viagra stupid
priligy choice – dapoxetine reason cialis with dapoxetine crystal
cialis soft tabs online inner – viagra super active pills mother viagra oral jelly online private
cialis soft tabs cast – tadarise pills realize viagra oral jelly envelope
brand cialis neither – brand levitra apparent penisole project
cenforce court – tadalis convince brand viagra online camp
brand cialis whip – brand levitra nail penisole copy
viagra professional online dollar – super kamagra mountain levitra oral jelly successful
priligy lucky – suhagra dusty cialis with dapoxetine fumble
rosuvastatin online shield – rosuvastatin pills advance caduet instrument
simvastatin pride – atorvastatin extend atorvastatin operate
order nitroglycerin – how to get nitroglycerin without a prescription order generic diovan 160mg
lopressor 100mg canada – order metoprolol 50mg generic adalat 30mg price
order digoxin 250mg online cheap – order lanoxin 250 mg pill order furosemide 100mg for sale
buy ketoconazole 200mg online cheap – purchase lotrisone generic itraconazole tablet
famciclovir 500mg sale – famciclovir brand valcivir 1000mg over the counter
buy cheap generic terbinafine – purchase grifulvin v generic griseofulvin cheap
buy rybelsus 14mg sale – buy desmopressin generic order desmopressin generic
glyburide 5mg us – glucotrol price order forxiga 10 mg pills
order desloratadine 5mg for sale – where can i buy beclomethasone buy albuterol generic
depo-medrol otc – buy fml-forte no prescription astelin 10ml tablet
purchase ventolin without prescription – buy promethazine without prescription theo-24 Cr ca
ivermectina 6mg – buy cefaclor generic cefaclor 250mg pill
amoxil without prescription – erythromycin over the counter purchase ciprofloxacin pill
augmentin order – ampicillin ca ciprofloxacin pill
generic atarax 10mg – pamelor tablet purchase amitriptyline without prescription
order quetiapine 100mg for sale – order ziprasidone 40mg without prescription order eskalith sale
clomipramine online – order citalopram how to buy doxepin
buy zidovudine – zyloprim 100mg oral
clozapine 50mg over the counter – buy generic frumil online pepcid 20mg over the counter
order glycomet 500mg sale – metformin 500mg usa lincomycin tablet
purchase lasix pill – prazosin sale captopril buy online
flagyl 400mg usa – oral cefaclor 250mg brand azithromycin
how to get acillin without a prescription buy acillin without prescription amoxicillin uk
order valacyclovir 500mg generic – buy cheap zovirax zovirax price
ivermectin 6mg stromectol – buy sumycin online cheap buy generic tetracycline 250mg
stromectol generic – where can i buy ciprofloxacin buy sumycin without prescription
order generic ciplox 500 mg – buy ciprofloxacin buy erythromycin sale
brand cipro 1000mg – brand keflex order augmentin 375mg for sale
cipro 500mg tablet – oral keflex 500mg augmentin 1000mg for sale
lipitor 80mg usa lipitor without prescription lipitor 20mg oral