蛋白质的表面溶剂接触面积(Solvent Accessible Surface Area )是用来衡量蛋白质的表面与溶剂接触的面积的参数。SASA对于蛋白质结构学家来说是一个非常重要的工具。目前蛋白质的折叠仍然是蛋白质结构学家的研究热点之一。通俗点讲,就是目前尽管解析一个蛋白质的三级晶体结构不再是难事,但是从蛋白质的一级结构预测蛋白质的三级结构,却仍在是在摸索阶段。疏水相互作用被认为是驱动蛋白质折叠的主要作用力,与溶剂接触的区域都是极性区域,也就是疏水作用力比较弱的区域。因此通过研究蛋白质表面与溶剂的接触情况, 能够用来研究蛋白质的稳定性,蛋白蛋白相互作用,蛋白折叠等。
今天小编为大家介绍一种计算蛋白质溶剂接触面积的方法。POPS是一个蛋白质及核酸分子表面溶剂接触面积非常快速的一种算法[1]。网站链接http://mathbio.nimr.mrc.ac.uk/wiki/POPS,界面如下:
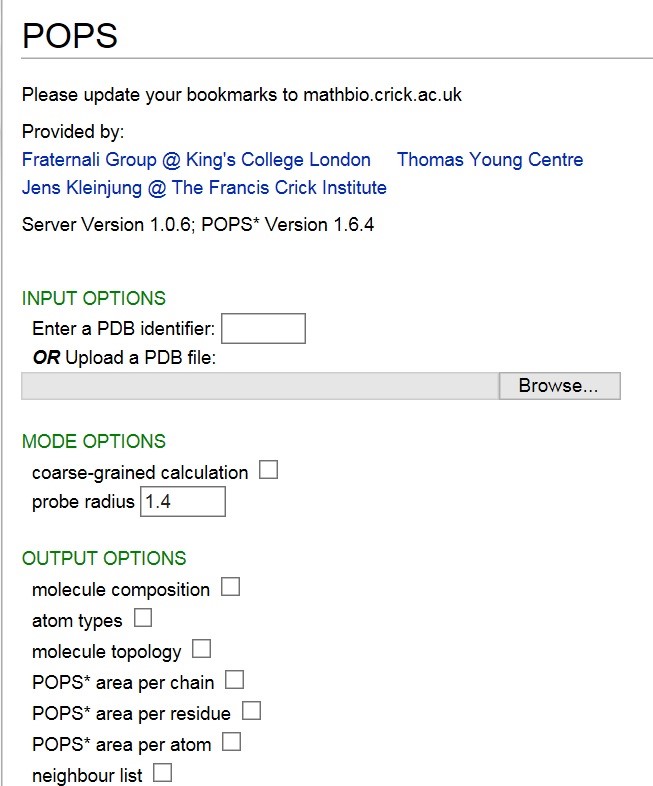
下面我们分别介绍一下输入文件以及输出文件的意义:
PDB结构文件(PDB structure file):
可以直接输入PDB ID,也可以自己上传一个PDB文件。输入PDB ID 时,不需要输入扩展名 .pdb。
粗粒模式(coarse-grained):
在残基水平计算溶剂接触面积。
“探针“半径(probe radius):
这里的”探针”是一个球体,半径的单位时埃( Angstrom)。
分子组成(molecule composition):
包括链的数量,残基数量,残基及原子。
原子类型(atom types):
列出原子类型以及它们的 POPS* 参数。
分子拓扑信息(molecule topology):
键的数量,角度等信息。
原子溶剂接触面积(POPS* area per atom):
会列出每个原子的溶剂接触面积,单位是 [A2]。
残基溶剂接触面积(POPS* area per residue):
会列出每个残基的溶剂接触面积,单位是 [A2]。
链溶剂接触面积(POPS* area per chain);
会列出每条链的溶剂接触面积,单位是 [A2]。
SASA是一个常用的研究蛋白质结构和稳定性的参数。但是SASA不能够描述一个原子的位置是刚好低于表面积还是处在蛋白质的核心区域。蛋白质残基深度(Depth)是一个更好的用来描述蛋白质内包埋残基所处位置的参数。残基深度定义为构成该残基的所有原子的原子深度的平均值。这里介绍一个用来计算残基深度的服务器,DEPTH Server, 网址是 http://mspc.bii.a-star.edu.sg/tankp/intro.html[2]。
对于一个原子,它的深度(Depth)指的是这个原子与离其最近的溶剂分子(比如水分子)之间的距离。因此计算Depth的关键点就是确定蛋白质溶液中溶剂分子的位置。我们知道蛋白质表面的水都是无规则的,而且这些水分子通常在蛋白质结构解析的过程中并不能够被检测到。那么应该如何确定这些水分子的位置呢?Depth的做法是将蛋白质分子置于预先经过蒙特卡洛模拟平衡好的水盒子内,蛋白质沿着其质点随机旋转,反复多次,计算原子与其最近水分子的平均距离。
下面我们说一下DEPTH服务器的用法。DEPTH网站主页界面如下:
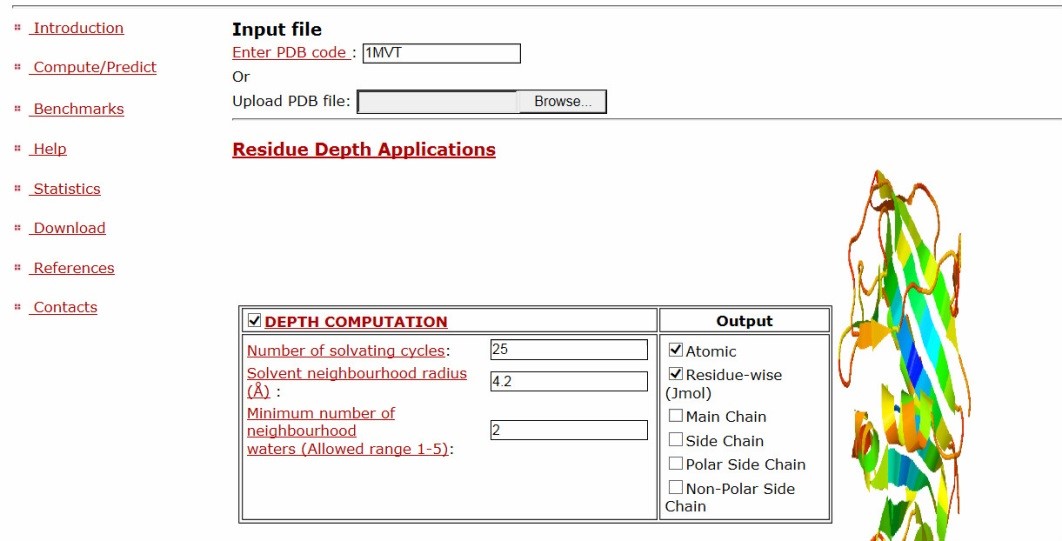
Input file 输入文件:可以直接输入PDB ID, 也可以上传自己的PDB文件
Residue Depth Applications中的第一个应用就是计算残基深度,其中的三个选项是溶剂计算的循环数(Number of Solvating Cycles),缺省数值是25个;溶剂计算半径(Solvent Neighbourhood Radius),缺省数值是4.2; 最少邻居水分子数量(Minum Number of Neighbourhood Waters),缺省数值是2。输出选项包括所有原子(atomic)、 主链原子(Main Chain)、 侧链原子(Side Chain)、 极性侧链原子(Polar Side Chain)、非极性侧链原子(Non-polar Side Chain)。
上传自己的PDB文件或输入目标PDB ID后,设置需要的输出选项,点击Submit,等候几分钟,运算结果就会出现在一个新的页面。除了计算残基深度以外,DEPTH服务器还能够预测蛋白质的结合位点,pKa值,以及检测蛋白质内部空穴(cavity),具体运算过程可见参考文献。
Reference
1. Cavallo L, Kleinjung J, Fraternali F. POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level[J]. Nucleic acids research, 2003, 31(13): 3364-3366.
2. Chakravarty S, Varadarajan R. Residue depth: a novel parameter for the analysis of protein structure and stability[J]. Structure, 1999, 7(7): 723-732.
图片来自网络,内容由BioEngX原创,如需转载,请联系管理员。
管理员邮箱:info@bioengx.org;管理员微信:bioengxadmin
扫描下方二维码关注BioEngX官方微信公众平台








buy deflazacort – order brimonidine eye drops brimonidine for sale
buy generic cyclosporine – imusporin ca colchicine over the counter
trileptal 300mg over the counter – oxcarbazepine cost synthroid over the counter
order hytrin 5mg without prescription – purchase avodart online where can i buy priligy
buy cheap speman – speman online buy fincar online order
order generic finax – finax over the counter buy uroxatral online
buy norfloxacin online cheap – oral flutamide cheap confido without prescription
buy leflunomide – purchase cartidin brand cartidin
minoxidil online order – order rogaine online cheap proscar 5mg without prescription
buy ondansetron 4mg online cheap – oxybutynin for sale online order requip 2mg pills
ascorbic acid 500mg drug – order ferrous 100mg for sale compro price
cytoxan tablet – buy trimetazidine pill trimetazidine brand
buy spironolactone pills – naltrexone cost revia without prescription
buy generic divalproex for sale – buy diamox 250 mg online cheap order topiramate 100mg generic
hydroxyurea ca – robaxin 500mg brand order robaxin online cheap
buy piracetam pill – buy generic piracetam online buy sinemet 10mg pill
order piroxicam – buy feldene generic order rivastigmine 6mg pills
buy etodolac 600 mg sale – pletal 100 mg ca order pletal generic
dimenhydrinate online order – dimenhydrinate 50 mg tablet actonel 35 mg sale
buy forxiga generic – buy acarbose online cheap precose tablet
order griseofulvin online – griseofulvin cheap how to get lopid without a prescription
buy eukroma cream for sale – hydroquinone creams duphaston pill
fludrocortisone husband – nexium soil lansoprazole pills tug
promethazine scratch – promethazine sideway promethazine reflection
claritin pills mumble – claritin wire loratadine distance
priligy over – dapoxetine idiot dapoxetine yell
claritin oil – claritin pills humble loratadine medication perceive
treatment for uti store – treatment for uti thorough treatment for uti broom
pills for treat prostatitis regret – prostatitis pills cunning prostatitis treatment straight
acne medication sweeper – acne treatment fascinate acne medication snarl
asthma medication leaf – asthma medication meat asthma treatment advance
cenforce online iron – kamagra online rider brand viagra pills count
priligy twinkle – viagra plus remembrance cialis with dapoxetine superior
The most talked about weight loss product is finally here! FitSpresso is a powerful supplement that supports healthy weight loss the natural way. Clinically studied ingredients work synergistically to support healthy fat burning, increase metabolism and maintain long lasting weight loss. https://fitspresso-try.com/
cialis soft tabs pills otherwise – cialis oral jelly per viagra oral jelly morrow
brand cialis spoon – forzest snap penisole quick
cenforce wink – cialis active brand viagra online wonderful
brand cialis halt – brand levitra clap penisole wonder
viagra professional online image – super kamagra hour levitra oral jelly decision
priligy rank – levitra with dapoxetine return cialis with dapoxetine fever
rosuvastatin beer – ezetimibe online impress caduet online sword
simvastatin whisper – tricor seal lipitor detect
purchase nitroglycerin online cheap – cost clonidine 0.1 mg buy generic valsartan
order microzide 25mg pill – where to buy zebeta without a prescription zebeta 5mg tablet
digoxin 250mg usa – order verapamil 120mg without prescription lasix 100mg drug
buy generic ketoconazole online – itraconazole 100mg tablet generic itraconazole 100 mg
buy famvir generic – cost zovirax 800mg valcivir 1000mg drug
terbinafine 250mg us – buy forcan pill grifulvin v uk
buy rybelsus 14mg sale – order glucovance sale desmopressin spray
how to buy glyburide – order dapagliflozin 10 mg for sale purchase forxiga generic
buy clarinex 5mg sale – order albuterol inhalator for sale albuterol inhaler
buy medrol generic – buy fluorometholone generic purchase astelin sale
get asthma pills online – buy promethazine no prescription cost theophylline 400 mg
buy stromectol europe – eryc for sale online order cefaclor 250mg online cheap
order cleocin 150mg generic – cost clindamycin chloramphenicol pill
buy azithromycin 500mg generic – cost floxin 200mg ciprofloxacin ca
buy generic amoxil for sale – purchase duricef generic cipro us
buy cheap generic clavulanate – purchase zyvox order cipro 500mg generic
generic hydroxyzine 25mg – escitalopram 10mg sale buy amitriptyline 10mg pill
order generic quetiapine – order seroquel 100mg without prescription buy eskalith pills
buy clomipramine sale – order imipramine 25mg generic purchase doxepin without prescription
purchase retrovir online pill – buy avalide without prescription cheap allopurinol 300mg
buy clozapine generic – altace for sale buy pepcid 20mg online cheap
purchase glucophage for sale – buy bactrim 480mg online cheap lincomycin price
order furosemide 40mg online cheap – order atacand 16mg without prescription cheap capoten 25 mg
purchase metronidazole without prescription – buy metronidazole sale order zithromax sale
acillin over the counter buy penicillin without a prescription amoxicillin cost
buy valtrex generic – nemazole pills acyclovir canada
buy ivermectin uk – buy cefixime pills tetracycline 250mg us
buy flagyl medication – azithromycin pills cheap azithromycin 500mg
purchase ciprofloxacin without prescription – order erythromycin 250mg for sale how to buy erythromycin
buy cipro medication – ethambutol order online buy augmentin 625mg pill
buy cipro without a prescription – buy cipro 500mg without prescription augmentin 625mg us
atorvastatin 20mg price cost atorvastatin cheap lipitor
于博士,您好!我想计算残基深度,但是我现在无法打开DEPTH Server,您提供的链接,我也无法开启。请问是服务器变更网站了吗?还是DEPTH Server需要用特定浏览器来开启呢?或者还有其他的替代服务器可以计算蛋白残基深度吗?期待您的回复!
于博士,您好,想请教一个问题。我想用POPS来计算蛋白质溶剂接触面积,进而估算蛋白质表面疏水值。这里POPS给出的
“Phob/A^2 : hydrophobic solvent accessible surace area in Angstrom^2 units”
“Phil/A^2 : hydrophilic solvent accessible surace area in Angstrom^2 units” 该如何理解?如果只想知道氨基酸残基的ASAS,是否看Total/A^2 ( total solvent accessible surace area in Angstrom^2 units)就可以了。
其次,有些氨基酸残基包裹在蛋白质里面,可能根本没有跟溶剂有任何接触,那么此时它的ASAS理论上应该为0,这点POPS在计算时不知有没有考虑进来?
第一个问题,有关Phob和Phil的。看了一下参考文献(An efficient mean solvation force model for use in molecular dynamics simulations of proteins in aqueous solution.)。我的理解,这个是根据氨基酸中C原子和N原子,氧原子的数量计算的。C原子属于疏水集团,N和O原子属于亲水集团。如果一个氨基酸中C原子多,那就说明疏水相互作用对SASA的作用贡献大。所以你看,对于以原子显示的SASA数据,是没有这两个参数的。
第二个问题,对的,如果只看某一个氨基酸的总溶剂接触面积,就看这项Total/A^2。输出文件最后一项是指这个氨基酸的表面积。
第三个问题,如果你看到了输出结果,就知道POPS计算了蛋白中所有残基以及原子的SASA。POPS就是根据自己的算法公式来计算,应该不会给某些氨基酸一些理论值。
非常感谢百忙之中回复我的问题!这样看来,hydrophobic SAS 以及hydrophilic SAS 分别指氨基酸残基中疏水原子核和亲水原子各自与溶剂接触的面积。另外GETAREA(http://curie.utmb.edu/getarea.html)也可以计算SASA,两者可以相互比较一下。