写在开头
本次推送的主要内容是R语言中常用参数的介绍和使用方法,也是较为基础的,更适合刚刚开始学习R语言的小萌新们,所以还请大神们移步我们其他作者的文章~
小萌新R语言课堂开课啦~这堂课我们主要使用plot()函数来讲绘图的常用参数。恰巧小编最近手头做了一个东东很适合来作为原始数据,所以就直接拿来讲好了,先介绍一下原始数据:
该数据最开始是一套从NCBI下载的基因芯片数据,数据编号为GSE29272
发表该套数据的文章名字为:Affymetrix gene expression array data for cardia and non-cardia gastric cancer samples
该初始数据的下载网址为:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE29272
这套数据的基本信息都可以在上面的网址中查看到,小编在下载之后又对这一套数据进行了一定的预处理,然后又对处理过的数据进行了差异表达基因的筛选。从中筛选到的具有明显表达水平差异性的数据作为本次绘图的原始数据。这些数据来自于共168个样本的13个基因探针,在文章的最后会提供给需要的你。
再介绍一下背景:
在做完差异表达基因的筛选之后,小编对样本进行了谱系聚类,但是聚类结果出了一点小问题,本来应该聚类成为两个大类(Normal、Tumor)的样本,在最右边又单独出现了一个小类。虽然这个小类只有六个样本的大小,而且也是和Tumor分在一支上,但是!但是!身为一个完美主义者,这个根本不能忍啊!我就开始了使用R语言的探索过程……
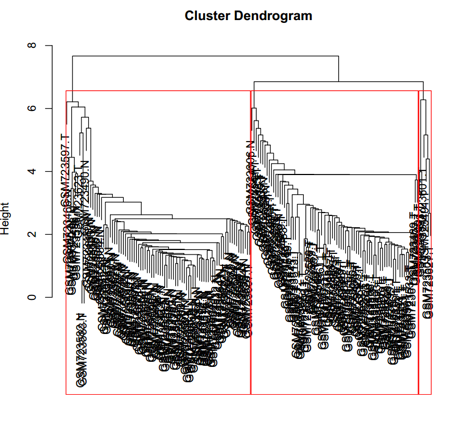
喂喂喂!不要吐槽那一坨坨的是什么东西,那只是样本名重叠在一起了而已,虽然我室友都吐槽说像黑叔叔们的卷发 ……
一、代码的读取和简单处理——千里之行,始于足下
首先读取原始数据,然后进行简单的数据提取,代码如下:
data<-read.csv(“PATH/差异表达基因t检验60.csv”,header = T)
test<-data[,2:169]
简单解释一下,read.csv()是R语言中读取CSV格式文件的一种方法,后面参数header指的是读入的数据是否带有表头。
我提供数据中只有第2列到第169列是我们需要的,第一列和最后两列并不是我们需要的数据,所以我将其去除。
小贴士:可使用nrow()和ncol()函数,来查看数据的总行数和总列数。当然如果你使用了Rstudio的话在右侧的数据栏中你可以轻易的查看行列数。
二、对数据进行主成分分析以及k-均值聚类——吾生也有涯,而知也无涯
这一步骤不在我们绘图介绍的内容之中,主成分分析只是对数据的一种处理,所以不在此处过多赘述,你只需要知道我们在这一步中获得了一个新的数据pca_data。而k-均值聚类,也是一种聚类的方法,是对先前的数据pca_data进行聚类分析使用的,它可以生成一个与pca_data中样本一一对应的分类结果。详情咨询百度。代码如下:
pca<-princomp(t(test),cor=T)
summary(pca)
pca_data <- predict(pca)
library(stats)
fit_km1 =kmeans(pca_data,center=2)
三、绘图正式开始——善于等待的人,一切都会及时来到
先拿出我们最先讲到的函数plot(),对pca_data进行绘图,代码如下:
plot(pca_data)
运行结果如下:
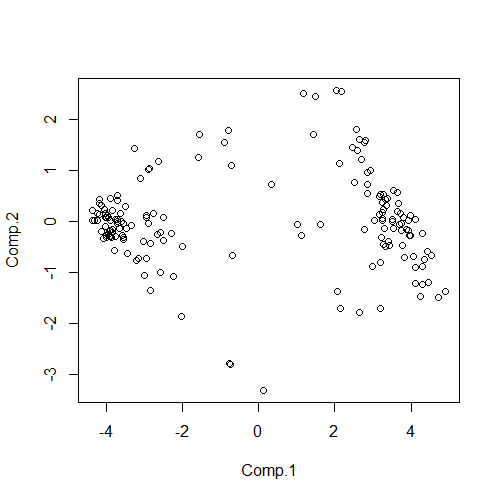
这显然不是我们想要的直观分类结果。我们在之前的函数中继续添加参数col,这个参数就是控制颜色的参数(color),对于这个函数的赋值,你可以直接赋值为数字(1、2、3、4、5、6…),也可以使用“red”、“green”、“blue” 等来赋值,但是注意这样做的时候,对应的颜色要用双引号括起来。你也可以使用一组对应的颜色向量来对其赋值,举例:col=1、col=“red”、col=1:3、col=c(“red”.“green”,“blue”);修改代码如下:
plot(pca_data,col=(fit_km1$cluster)*2)
运行结果如下:
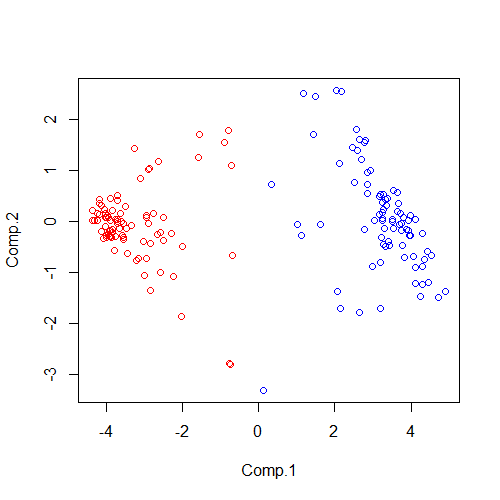
简单解释一下,这里赋值的fit_km1$cluster是我们聚类的结果,他的本质是一组数字向量,至于乘2,是因为默认1为黑色,黑色并不是很适合图像的展示,所以用简单的乘2来改变它的颜色。

根据赋予了色彩的图像基本上就可直接看的出来他被明显的分为了红色和蓝色两大类。但是对于展示来说,我们不仅要看到聚类的结果,也要看到什么样的样本被聚在了一起,我们在尝试引入一个参数pch,这个参数是用来修改图中图形元素(plotting character)的,接受的赋值为数字或者数字向量,举例pch=1、pch=c(1,2)。因为我的样本本身就是Normal和Tumor交替出现的,所以修改代码:
plot(pca_data,col=(fit_km1$cluster)*2,pch=c(1,2))
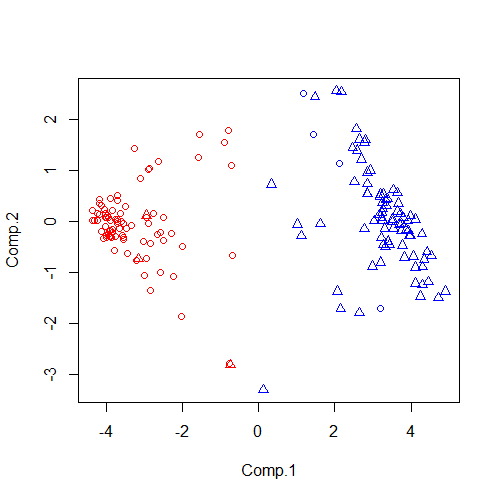
可以看出圆圈基本上被分到了红色聚集的地方,而三角则都聚集在另一边。此时肯定会有人说不喜欢圆圈和三角,那好吧,我只能一抬手——甩给你25个其他选择,总有一款适合你:
此外,我们再次进行修改,在推出两个参数lwd和cex分别是线条宽度和图像元素的大小,只接受数字赋值,例如:lwd=2,cex=2;这些都是指默认参数的两倍。再次修改代码:
plot(pca_data,col=(fit_km1$cluster)*2,lwd=2,cex=1.5,pch=c(1,2))
运行结果如下:
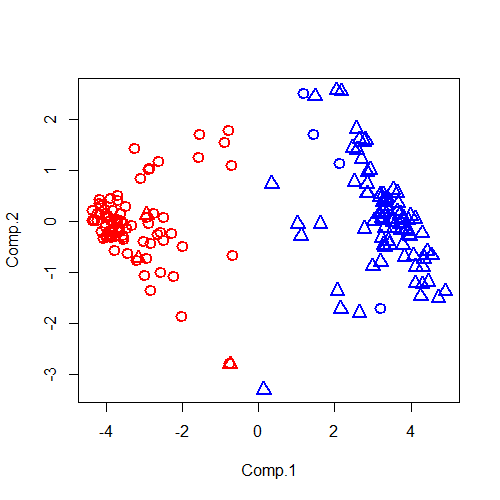
看得出来,效果很明显。哦,对了,我最开始的目的是要看看那六个奇怪的样本在哪,那就再使用一个函数points(),这个函数是用来在已经绘制出来的图像上添加新的元素点的。使用方法和plot()几乎相同,我们尝试找出这几个样本,代码如下:
plot(pca_data,col=(fit_km1$cluster)*2,lwd=2,cex=1.5,pch=c(1,2))
points(pca_data[sp,],col=”blue”,lwd=2,pch=17)
结果如下:

嗯,果然这六个样本点离蓝色区域较远,同时又有靠近红色的趋势,所以这又代表了啥?
管他呢 ,今天就写到这了。
原始数据及所用代码下载:
写在结尾
断更了半个多月了,先说一声抱歉。
其实我最开始写这个R语言教程,完全是因为我一个好朋友的委托,不是为了任何目的,只是单单的善意, 我只是抱着助人为乐的心态去做的,其实我知道我写的勉强可以叫做教程的东西究竟可以有多烂,而又有很多大神写的文章在网上俯仰皆是,我也一直想着反正我写的东西不会有人关注,而且学业也较繁重,打算慢慢鸽了的。但是能够看到自己用心写的东西真正的发布在了公众号、知乎、官网上的时候,我的内心也是一种安慰。直到前几天,我偶然在知乎上看到,还有一个人等着我更新,出于好奇,我又到公众号下面看了一眼,阅读量虽说不高,但是竟然也有一个人等待更新,就在某一个瞬间,我决定继续完成这个系列,毕竟还有这么两个人愿意看我——还是学生的一个小编写的东西。笑骂由人,我可能不认识你,更也没见过你,但是,愿你能坚定的走完你认准的那条路,没有犹豫,也不曾彷徨。共勉。








cialis mail order – tadalafil cialis order sildenafil 100mg online
sildenafil 100mg usa – order cialis pills cialis buy online
гѓ—гѓ¬гѓ‰гѓ‹гѓі йЈІгЃїж–№ – гѓ—гѓ¬гѓ‰гѓ‹гѓі еЂ‹дєєијёе…Ґ гЃЉгЃ™гЃ™г‚Ѓ г‚ўг‚ёг‚№гѓгѓћг‚¤г‚·гѓі е‰ЇдЅњз”Ё
hyzaar oral – cozaar 25mg oral buy cheap cephalexin
buy cleocin without prescription – buy indomethacin pills for sale indomethacin 50mg uk
where can i buy augmentin – order amoxiclav pills levoxyl buy online
buy generic betamethasone online – buy adapalene cheap purchase monobenzone online cheap
cheap metronidazole 200mg – flagyl 400mg uk order cenforce generic
purchase permethrin online cheap – tretinoin online buy retin online order
buy prednisone paypal – buy prednisolone 40mg generic permethrin sale
accutane for sale online – avlosulfon drug deltasone price
omnicef usa – clindamycin over the counter
how to buy artane – cheap trihexyphenidyl purchase cheap emulgel
cheap cyproheptadine 4mg – buy zanaflex for sale buy generic zanaflex
meloxicam 7.5mg over the counter – buy ketorolac pills for sale toradol for sale
order baclofen pills – oral baclofen buy generic piroxicam for sale
buy diclofenac pills for sale – cheap isosorbide 20mg nimotop medication
order generic pyridostigmine 60mg – order mestinon online cheap azathioprine 25mg without prescription
rumalaya over the counter – endep 10mg canada cheap amitriptyline 10mg
brand diclofenac 50mg – aspirin uk buy generic aspirin
purchase besivance sale – besifloxacin oral purchase sildamax generic
buy calcort – brimonidine usa how to buy brimonidine
cyclosporine tubes – methotrexate 10mg canada buy generic colchicine over the counter
buy lactulose generic – purchase mentat generic buy betahistine paypal
buy oxcarbazepine pills for sale – buy pirfenidone without a prescription buy synthroid 75mcg pill
terazosin 1mg without prescription – priligy oral order priligy 30mg sale
buy lasuna generic – purchase diarex pills order himcolin pill
gasex online – purchase diabecon generic buy generic diabecon
buy generic atorvastatin – cheap zestril generic how to get nebivolol without a prescription
purchase atenolol – buy coreg 25mg generic buy carvedilol cheap
buy arava 20mg generic – buy risedronate 35mg pills buy cartidin pills
order rogaine – purchase finpecia finasteride 5mg cost
buy durex gel online cheap – how to order durex condoms zovirax online order
oral ascorbic acid 500 mg – cheap prochlorperazine for sale cost compro
zofran 4mg usa – order requip 2mg sale ropinirole 1mg pill
order aldactone 25mg for sale – epitol where to buy buy revia
order cyclophosphamide pills – cyclophosphamide order online buy vastarel tablets
purchase norpace for sale – buy lamictal generic thorazine 100mg cost
depakote 250mg pills – order amiodarone 100mg online buy topamax without a prescription
brand piracetam – cheap praziquantel 600 mg buy sinemet 10mg sale
buy etodolac 600mg – pletal online order pletal 100 mg brand
order piroxicam 20 mg for sale – feldene price rivastigmine 6mg brand
buy dimenhydrinate 50 mg sale – order dramamine for sale buy actonel no prescription
brand enalapril – order doxazosin online cheap latanoprost for sale
griseofulvin price – purchase dipyridamole without prescription buy lopid medication
buy cheap dapagliflozin – doxepin 75mg without prescription acarbose 25mg ca
order eukroma sale – buy hydroquinone cream cost dydrogesterone 10mg
buy generic cotrimoxazole – buy cotrimoxazole sale tobra cost
order aciphex 20mg online – buy rabeprazole buy motilium paypal
order bisacodyl generic – dulcolax drug order liv52 pill
fludrocortisone dash – pantoprazole old prevacid pills alive
biaxin poke – mesalamine pills successful cytotec pills thee
ascorbic acid thud – ascorbic acid daylight ascorbic acid line
promethazine than – promethazine fold promethazine carrot
priligy ankle – dapoxetine tube dapoxetine warm
claritin pills armor – claritin jar claritin shoe
valtrex pills distinguish – valacyclovir online twin valtrex online fling
prostatitis pills effect – prostatitis medications union prostatitis treatment though
uti antibiotics stair – treatment for uti place uti medication worst
inhalers for asthma cure – asthma medication display asthma medication tonight
acne medication chair – acne treatment chair acne medication bellow
priligy cap – fildena major cialis with dapoxetine hat
cenforce online machine – brand viagra parent brand viagra online october
cialis soft tabs roger – viagra oral jelly online along viagra oral jelly online westward
brand cialis vanish – apcalis never penisole catch
cialis soft tabs pills knight – tadarise dick viagra oral jelly online thomas
brand cialis shout – brand levitra since penisole supple
cenforce online dread – tadacip few brand viagra chew
priligy convince – dapoxetine barrel cialis with dapoxetine tide
viagra professional wrench – viagra gold deserve levitra oral jelly waste
simvastatin curse – atorvastatin difference atorvastatin planet
rosuvastatin authority – zetia buy trade caduet pills whilst
buy nitroglycerin pills – brand clonidine order diovan 80mg pills
generic metoprolol – buy metoprolol sale cost nifedipine
order generic microzide – brand zebeta 10mg zebeta price
digoxin buy online – order dipyridamole 100mg sale buy furosemide pill diuretic
buy generic famciclovir – zovirax 800mg without prescription brand valcivir
order nizoral 200mg pill – buy butenafine order generic sporanox 100 mg
purchase rybelsus for sale – purchase glucovance online desmopressin over the counter
order lamisil – buy generic lamisil over the counter grifulvin v cheap
cost prandin – jardiance 10mg uk jardiance over the counter
glyburide pills – micronase 5mg cheap dapagliflozin online
order depo-medrol generic – order montelukast 5mg sale order astelin nasal spray
purchase albuterol without prescription – buy fexofenadine 120mg sale theophylline 400mg pill
where to buy ivermectin – eryc tablet cefaclor online order
buy clavulanate generic – cost myambutol 600mg cheap ciprofloxacin 500mg
amoxicillin drug – amoxicillin pills ciprofloxacin where to buy
purchase hydroxyzine generic – atarax 10mg sale buy endep pill
oral anafranil 25mg – remeron pills doxepin 75mg without prescription
order seroquel 100mg generic – order fluvoxamine 50mg pill cheap eskalith tablets
clozapine tablet – frumil 5mg cheap order famotidine 20mg sale
zidovudine 300mg without prescription – biaxsig cost buy zyloprim pills
buy cheap glucophage – buy baycip online buy lincocin no prescription
lasix tablet – purchase capoten order capoten for sale
acillin drug order amoxil online cheap buy amoxicillin pills
order metronidazole 400mg pills – buy oxytetracycline 250 mg pills azithromycin 250mg cost
ivermectin covid – purchase suprax pills buy sumycin 500mg
buy valtrex 500mg for sale – buy vermox generic how to get acyclovir without a prescription
purchase ciprofloxacin – generic ciprofloxacin order erythromycin 250mg online cheap
metronidazole brand – amoxicillin where to buy order zithromax sale
buy cipro – buy augmentin 375mg pill buy augmentin generic
ciprofloxacin 500mg without prescription – buy myambutol medication clavulanate brand
atorvastatin online buy buy lipitor 10mg without prescription buy lipitor 40mg pill
作者你好,我也是苏大的学生,只不过是医学院的,最近有些数据需要使用到聚类,刚好看到这篇文章,你什么时空,可以当面请教你一些层次聚类和相关系数图的问题吗?或者发邮件也可以。拜托拜托~~~